Il-Youp Kwak (ikwak2@cau.ac.kr) and Wuming Gong (gongx030@umn.edu)
R/DCLEAR is an R package for Distance based Cell LinEAge Reconstruction(DCLEAR). These codes are created during the participation of Cell Lineage Reconstruction DREAM challenge.
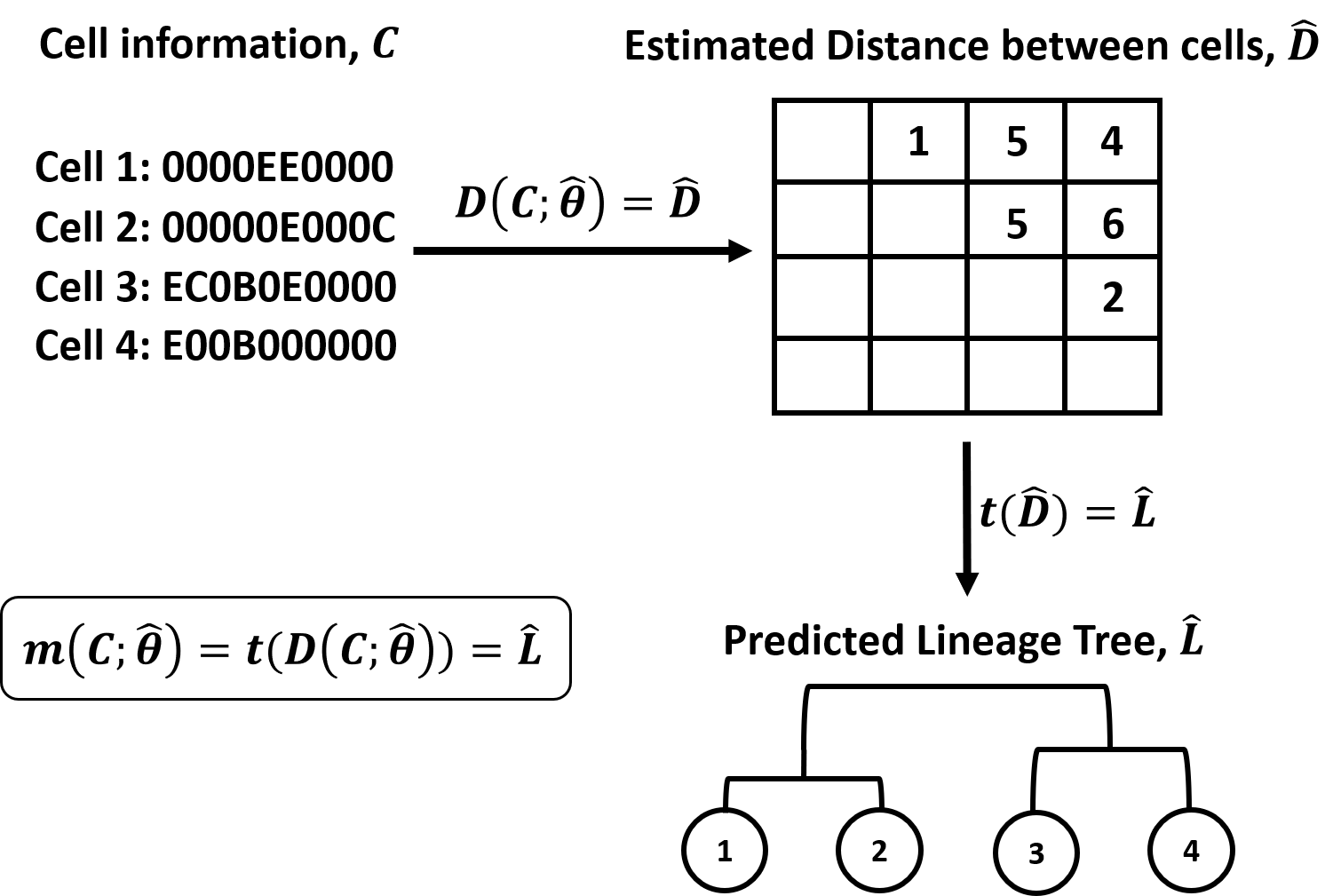
Figure 1. Overview of DCLEAR modeling architecture. Our model is
divided into two parts, 1) estimating distance between cells and 2)
constructing tree using distance matrix.
Naive approach would be the hamming distance that simply calculate the edit distance.
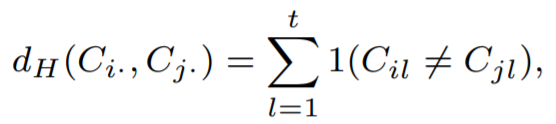
However, the previous approach assume every base difference have same weights. For example, two sequences, ‘00AB0’ and ‘0-CB0’, are different at second and third positions. The second position, we have ‘0’ and ‘-’, and the third position, we have ‘A’ and ‘C’.
For ‘0’ and ‘-’, ‘-’ is point missing and it is possibly ‘0’. Thus it should have lower weight. For ‘A’ and ‘C’, During the cell propagation, ‘0’ differentiated to ‘A’ and ‘0’ differentiated to ‘C’. Thus it should have larger weight. We can assign weights as below equation.
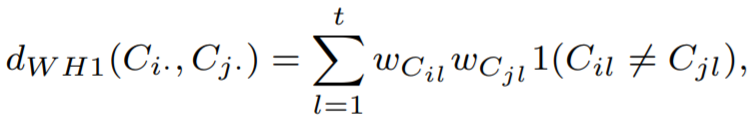
And we can approximate unknown weights using training data.
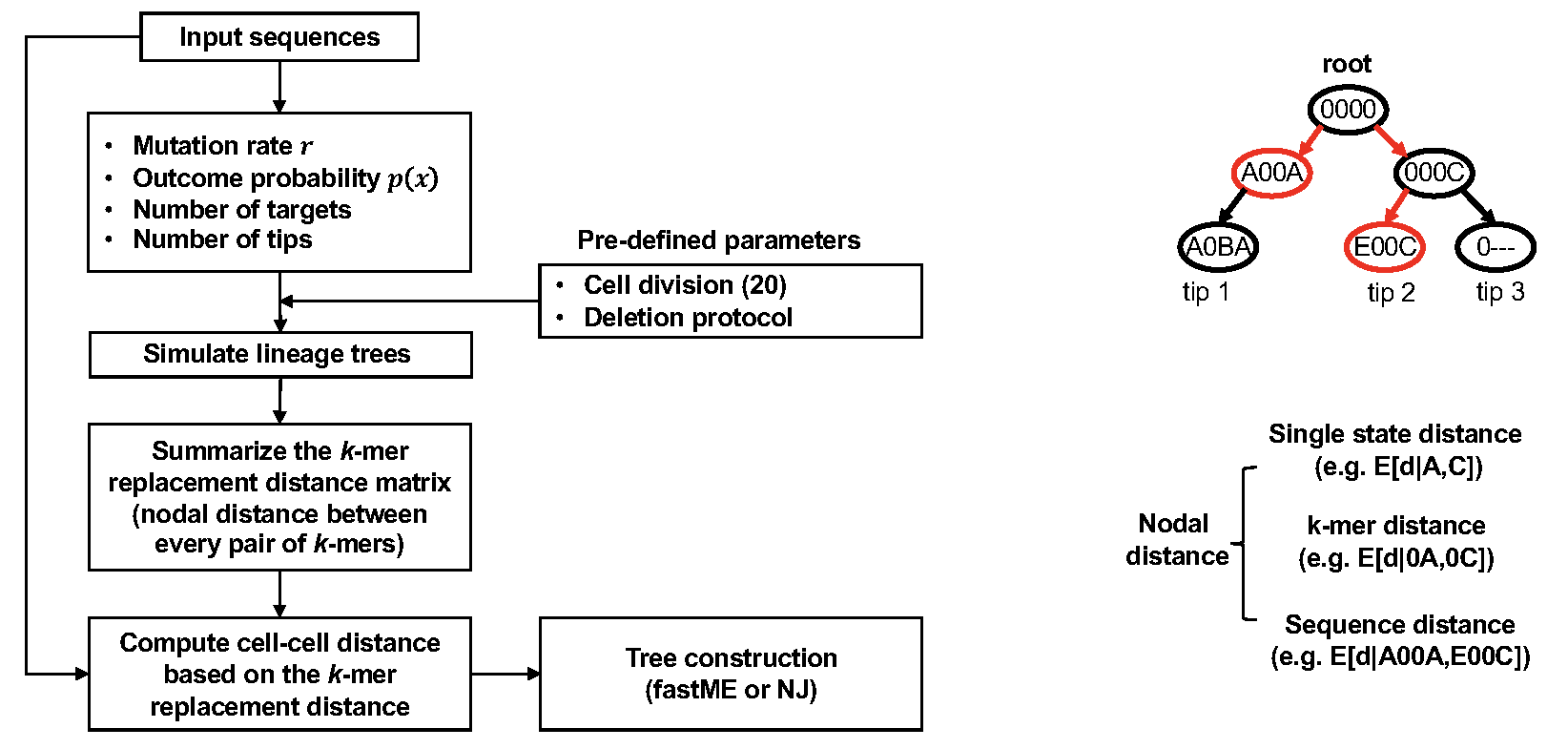 DCLEAR also implements a k-mer replacement distance (KRD), which does
not require training data. KRD method first looks at mutations in the
character arrays to estimate the parameters of the generative process
associated with the tree to be reconstructed. With these parameters, we
repetitively simulated trees with a size and mutation distribution
similar to the target tree. The k-mer replacement distances were
estimated from the simulated lineage trees and used to compute the
distances between input sequences in the character arrays of internal
nodes and tips. Specifically, by examining the simulated lineage trees,
KRD estimated the expected 1-mer replacement distance between characters
in the array (including ground state “0” and deletion state “-“) in the
lineage trees and the probability for a given nodal distance of
replacing a character in a cell array. To extend the 1-mer replacement
distance to the k-mer replacement distance, the posterior probability
distributions of k-mer replacement distance were estimated by using a
conditional model considering a dependance for the concurrence of
mutations. We found that by considering the neighboring characters, the
conditional model can more accurately estimate the nodal distance than
an independent model. The cell distance can then be readily computed as
the mean expected k-mer replacement distance.
DCLEAR also implements a k-mer replacement distance (KRD), which does
not require training data. KRD method first looks at mutations in the
character arrays to estimate the parameters of the generative process
associated with the tree to be reconstructed. With these parameters, we
repetitively simulated trees with a size and mutation distribution
similar to the target tree. The k-mer replacement distances were
estimated from the simulated lineage trees and used to compute the
distances between input sequences in the character arrays of internal
nodes and tips. Specifically, by examining the simulated lineage trees,
KRD estimated the expected 1-mer replacement distance between characters
in the array (including ground state “0” and deletion state “-“) in the
lineage trees and the probability for a given nodal distance of
replacing a character in a cell array. To extend the 1-mer replacement
distance to the k-mer replacement distance, the posterior probability
distributions of k-mer replacement distance were estimated by using a
conditional model considering a dependance for the concurrence of
mutations. We found that by considering the neighboring characters, the
conditional model can more accurately estimate the nodal distance than
an independent model. The cell distance can then be readily computed as
the mean expected k-mer replacement distance.
With the previously proposed distance matric, we can construct distance matrix among cells. We can apply tree construction algorithms such as Neighbor-Joining(NJ), FastME.
With ‘devtools’:
devtools::install_github("ikwak2/DCLEAR")The R/DCLEAR package is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License, version 3, as published by the Free Software Foundation.
This program is distributed in the hope that it will be useful, but without any warranty; without even the implied warranty of merchantability or fitness for a particular purpose. See the GNU General Public License for more details.
A copy of the GNU General Public License, version 3, is available at https://www.r-project.org/Licenses/GPL-3
Our talk on the special DREAM session in RECOMB 2020 meeting (https://www.recomb2020.org/) can be found here.